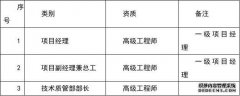
临床试验操作流程
时间:2026-01-18
时间:2026-01-18
一、试验启动阶段:
l 收集该药物已有的各种资料
理化性质、药理、药化、药效、毒理以及已有的临床研究资料。
l 制作研究者手册
研究者手册是临床试验开始前的资料汇编。研究者手册的内容一般包括:目录、序言、化学和物理性质、临床前研究、药理学研究、药代动力学研究、毒理学研究、已有临床资料、药品使用信息。
l 筛选主要研究者
①从临床基地名录中筛选医院,然后选择合适的主任级医生。
② 联系主任医生,约定拜访时间
③携研究者手册拜访
④初步交谈,如其有意,则在让其签署保密协议之后,请他阅读研究者手册
⑤再次拜访,与其讨论方案、试验时间、试验费用等问题
⑥如此拜访2-3位主任医生,从中选择最佳人选
⑦与选定的主要研究者签订合作协议
⑧试验文件准备
⑨会同CRO、申办者、主要研究者,共同制定临床试验方案、病例报告表、知情同意书样本、原始文件
⑩CRO制定临床试验中其他用表
l 其他研究者筛选
①从临床基地名录中选择其他可能的研究者,人数为最终要求人数的120%
②准备资料:方案、研究者手册、知情同意书样本、研究者责任书、保密协议
③ 逐一拜访
④ 如有意向,在其签署保密协议后,请其阅读文件
⑤ 与其谈论方案,要求提供其医院所能提供的病例数、时间进度和经费预算
⑥ 选定合适研究者,取得其医院管理部门的批准证明
l 取得所有参加医院的伦理委员会批件
①伦理委员会将对有关批件、药检报告、研究者手册、知情同意书样本、试验方案、病例报告表进行审批
②报省级药品监督管理部门备案
l 试验药品准备
①敦促申办方进行试验用药品的送检
②生物统计师设计随机分组方案
③ 根据随机分组方案,设计药品标签
④设计应急信件
⑤药品包装:为每一个受试者准备好一盒药物,药盒上贴好标签,并装入相应的应急信件 l 召开研究者会议
① 确定试验方案
②确定时间进程、病例数、预算
③各方签订协议
l试验人员培训,以达到统一标准的目的
l试验相关文件、表格、药品分发到各研究者
l致函省级、国家药监局,致函申办者
l启动临床试验
二、临床试验进行阶段:
l 制定试验的总体访视访视时间表
l 每一次访视前,回顾试验的进展情况、前次未解决的问题
l 与研究者联系,确定访视日期,并了解试验用品是否充足
l 制定本次访视工作的计划、日程表,准备访视所需的文件资料和物品
l 与研究者会面说明本次访视的主要任务,了解试验进展情况(受试者入选情况、病例报告表填写情况),以前访视所发现问题的解决情况
l 核对并更新研究者管理文件册,检查并补充试验用品
l 检查知情同意书(注意版本、签名及日期)
l 核查原始文件及病例报告表(注意对试验方案的依从性、完整性、一致性、严重不良事件的发现与报告)
l 收集病例报告表
l 试验药品的核查(存放情况、发放回收情况记录、清点药品并与相应记录核对、检查盲码信封、使用是否违反方案要求)
l 记录所发现的问题
l 与研究者一起讨论和解决此次访视发现的问题,交流其他研究单位的进展和经验
l 将取回的药品、物品、已签署的知情同意书、病例报告表等按规定存放
l 填写访视报告
l 更新各项记录表格
l 对发现问题的追踪及解决
l 安排后续访视计划
l 临床试验过程中,如试验方案、知情同意书、病例报告表发生改动,需报伦理委员会审批 l 临床试验中发生严重不良事件,必须在24小时内报告药品监督部门,并尽快报告伦理委员会和申办者
l 病例报告表收集同时,生物统计师建立数据库,设定逻辑校验程序,并将收集到的病例报告表输入。输入过程中发现病例报告表有问题,则立即提交监察员,由监察员在下次访视中加以解决。 l 当数据库中已有一定病例记录时,生物统计师开始编撰统计分析程序,并利用已有数据进行调试
三、试验总结阶段:
l 检查并解决常规访视中遗留问题
l 收集所有病例报告表并与原始文件核对检查
l 回收所有未使用药品,核对药品发放、使用、回收记录是否吻合
l 回收所有试验用品
l 更新所有记录表格
l 数据入库
① 在进行阶段,已经进行了一遍数据输入,收集所有病例报告表后,再输入一遍
② 将两遍输入的数据进行自动校对,输出两者差别表
③根据两者差别表,对照原始病例报告表进行改正
l 生物统计师编写数据逻辑校验程序,以程序分析数据库中数据的合理性
l 对于逻辑校验程序发现的问题,对照原始病例报告表。如果是输入错误,则加以改正;如果是原始病例报告表填写有误,则再返回医院,进行检查更正。
l 所有数据通过数据逻辑校验程序的审查后,锁定数据库。
l召开四方参与的盲态核查会议,检查应急信件及盲底,确认分析集
l 统计分析
①生物统计师(甲)编写统计分析程序
②对每个医院进行分析;对所有医院总和进行分析;对符合方案集进行分析;对意向集进行 …… 此处隐藏:84字,全部文档内容请下载后查看。喜欢就下载吧 ……